What is Disease-related Variant Annotation (DVA)?
DVA is a tool for predicting the functional impact of single nucleotide variants (missense) in the human genome.
In the past decade, single nucleotide variants (SNVs) have been identified as having a significant relationship with the development and treatment of cancer. Among them, prioritizing missense variants for further functional impact investigation is an essential challenge in the study of common disease and cancer. Although several computational methods have been developed to predict the functional impacts of variants, the predictive ability of these methods is still insufficient in the mendelian and cancer missense variants. Thus, we present a novel prediction method called the disease-related variant annotation (DVA) method that predicts the effect of missense variants based on a comprehensive feature set of variants, notably, introduce the allele frequency and protein-protein interaction (PPI) network feature based on graph embedding. Benchmarked against datasets of single nucleotide missense variants, the DVA method outperforms the state-of-the-art methods by up to 0.473 in the AUROC. The results demonstrate that the proposed method can accurately predict the functional impact of single nucleotide missense variants and substantially outperform existing methods.
In this article, two factors may contribute to the improved prediction performance. First, two new types of features have been introduced to significantly improve the predictive ability of this algorithm: 1) variant allele frequencies in different populations. When a SNP is widely present in the population, it often has no pathogenic effect on the operation of the organism. On the contrary, if a SNP only appears in a few individuals, its impact on the organism may be more pathogenic. It has been less taken into account in previous prediction tools. 2) PPI network features. Whether it is normal organic operations or harmful molecular changes, it is often not a single factor that promotes its development, but the interaction of multiple molecules or changes in key molecules that lead to essential changes in the entire working mechanism. In the past, little attention has been paid to the interaction mechanism of different molecules in the research on the effect of variants. Here we used the PPI network to represent the interaction of molecules with different variants through feature extraction and used these features to predict the impact of variants and achieved good performance. Second, a random forest model was constructed to predict the functional impact of variants. By merging the different types of features and the DVA model, the DVA algorithm has significantly improved the prediction of the impact of missense variants.
Methods - DVA (Disease-related Variant Annotation)
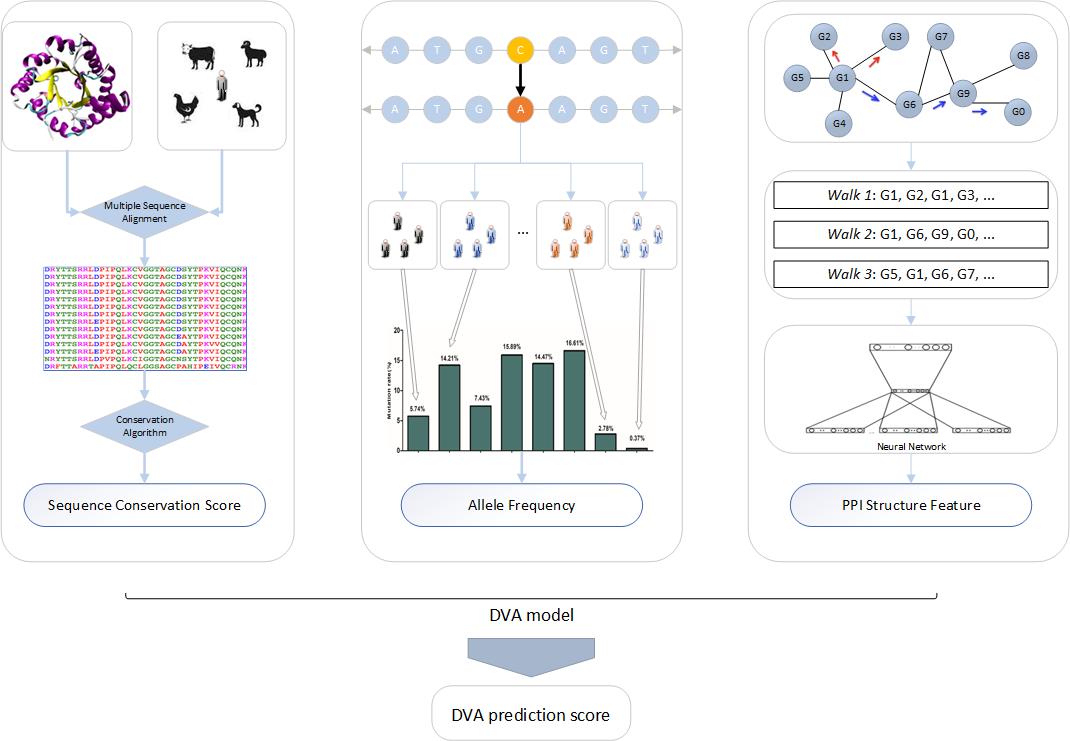
Figure 1. The overview of the DVA algorithm.
As shown in Figure 1, the DVA algorithm is mainly composed of a feature set and the DVA model. Among them, the feature set is mainly divided into three categories: sequence conservation, allele frequency, and the network structure of the protein (gene).
1. Sequence conservation The conserved elements include the vertebrate alignment and conservation (Phast) results, the genomic evolutionary rate profiling (GERP++) results and phastCons-Xway and phyloP-Xway conservation scores; The conservation of TF includes TFBS ChIP-Seq annotation dataset from the ENCODE project; 2. Allele frequency Complex diseases may be determined by low-frequency genetic variation. Thus, we incorporated the allele frequency data from the Genome Aggregation Database (gnomAD version v2.1.1), such as AF_male, AF_female, AF (all), AF_afr (African/African-American), AF_sas (South Asian), AF_amr (Latino/Admixed American), AF_eas (East Asian), AF_nfe (Non-Finnish European), AF_fin (Finnish), AF_asj (Ashkenazi Jewish), AF_oth (Other) and so on. 3. Network structure When a variant occurs in a key gene of the PPI network, its impact is generally greater than that of other genes. Therefore, we hope to introduce the characteristics of the PPI network into the prediction of SNP function effects to improve accuracy. However, the whole PPI network has a higher structural dimension, which will undoubtedly increase the computational complexity and because the network is sparse, it will affect the prediction work. Therefore, we use node2vec to obtain the graph embedding of PPI network (k dimension) as the network features. Finally, We used a random forest model, a machine learning technique, to predict whether a missense variant is disease-related or neutral.
The significance of DVA algorithm
Precision medicine and targeted therapy are active areas of disease research. The key to common and complex disease therapy is to understand which single nucleotide missense variants play a critical role in the development of the disease. With DVA, we describe a generic and effective framework for identifying the functional effect of single nucleotide missense variants based on multiple biological, computational, and network features. We demonstrate that the performance of DVA is much better than the state-of-the-art prediction tools using human missense variants. Meanwhile, we observed the general robustness of the DVA and it will be innovative for the study of the functional mechanism and impact of SNPs.
Reference
DVA: predicting the functional impact of single nucleotide missense variants